R software package to fit sparse additive interaction models with the strong heredity property. Interactions are limited to a single exposure or environment variable. The following figure (based on simulated data) gives an idea of the situation our method is trying to capture:
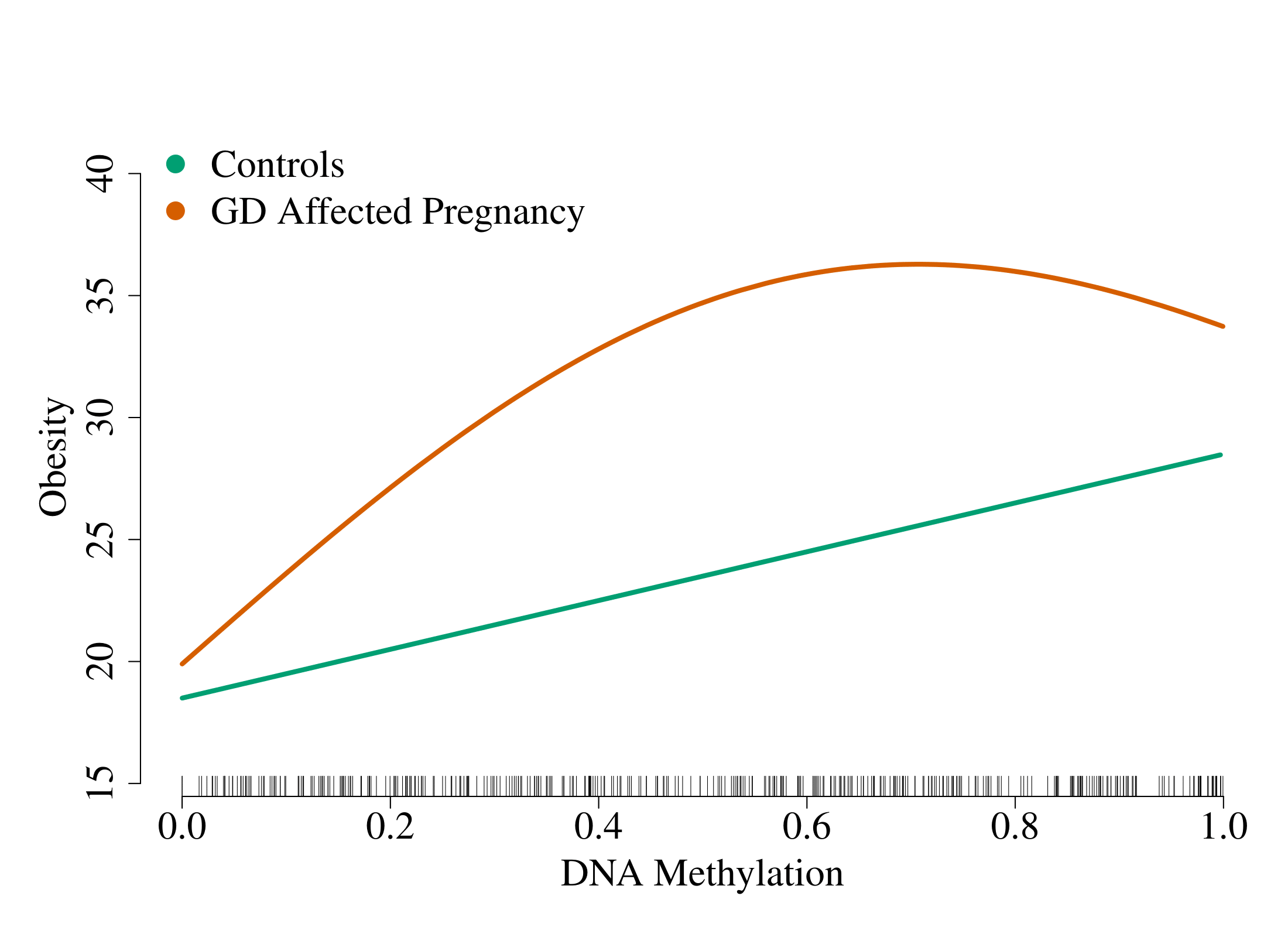
The x-axis is a predictor, the y-axis is the response and each line represents an exposure status. In this example, we see that the effect of the DNA methylation (at one CpG site) on obesity is linear for controls (unexposed: E=0) and non-linear for individuals with a gestational diabetes-affected pregnancy (exposed: E=1).
Installation
You can install the development version of sail from GitHub with:
Vignette
See the online vignette for details about the sail model and example usage of the functions.
Model Input
This method requires four inputs (let n be the number of observations and p the number of X variables):
-
X: n x p matrix of covariates. Can be high-dimensional, i.e., p >> n. Can also be continuous, categorical or a combination of both.
-
Y: a continuous response of length n
-
E: an exposure variable of length n. Can be continuous or categorical.
- A basis expansion function f(X_j) to be applied to each column of X, for example
Model Output
The sail method will search for all main effects and interactions between E and f(X_j) that are associated with the response Y, in a multivariable regression model. It will perform simultaneous variable selection and estimation of model parameters, and return a sparse model (i.e. many parameters with coefficient 0).
Contact
- Issues: https://github.com/sahirbhatnagar/sail/issues
- Pull Requests: https://github.com/sahirbhatnagar/sail/
- e-mail: sahir.bhatnagar@gmail.com
Latest news
You can see the most recent changes to the package in the NEWS.md file
Code of Conduct
Please note that this project is released with a Contributor Code of Conduct. By participating in this project you agree to abide by its terms.
